

aus dem Paracelsus Magazin: Ausgabe 5/1998
Hepatische Encephalopathien – Neuropsychatrische Symptome

Schlafstörungen als Korrelate einer Stoffwechselstörung von Melatonin im Rahmen subklinischer Leberfunktionsstörungen
Die hepatische Encephalopathie ist ein klinisches Syndrom, das bei schweren akuten und chronischen Lebererkrankungen auftreten kann. Es kommt dabei zu funktionellen, potentiell reversiblen Störungen der Gehirnfunktion. Die Symptomatologie ist charakterisiert durch Funktionsstörungen des Zentralen Nervensystems mit Beeinträchtigung intellektueller Funktionen, der Persönlichkeit und des Bewußtseins sowie EEG-Veränderungen. In 90 % der Fälle besteht eine Hyperammoniämie.
Das Spektrum neuropsychiatrischer Symptome einer hepatischen Encephalopathie reicht von kaum erkennbaren leichten Veränderungen von Gehirnfunktionen über psychiatrische und neurologische Symptome bis zum tiefen Koma. Die präzise Pathogenese der hepatischen Encephalopathie ist unbekannt. Es existiert keine kausale Therapie. Die subklinische hepatische Encephalopathie SHE, die oft bei Patienten mit Leberzirrhose zu finden ist (30-70 %),weist keine klinische Signifikanz auf. Psychometrische Tests geben jedoch Hinweise auf eine SHE. Veränderungen der Aufmerksamkeit und Bewegungsstörungen sind die häufigsten neurocognitiven Defizite. Schlafstörungen treten häufig bei Patienten mit Leberzirrhose auf. Sie sind charakterisiert durch Veränderung des Schlaf-Wach-Rhythmus, Schlaflosigkeit in der Nacht und Somnolenzam Tag .
Neben den klassischen Theorien bezüglich der Pathogenese der hepatischen Encephalopathie lassen sich auch Störungen des Vitamin-B12- und Biotin-Stoffwechsels infolge Abnahme der hepatischen Speicherbarkeit als Auslöser einer Hyperammoniämie in Erwägung ziehen.
 Die Problematik zentralnervöser Funktionsveränderungen infolge alkohol- und schadstoffinduzierter Leberschädigungen gewinnt unter den Schätzungen der Deutschen Hauptstelle für Suchtgefahren, nach der ca. 5 % aller Bundesbürger suchtkrank sind und 6,6 Millionen Bundesbürger einen problematischen Alkoholkonsum pflegen, nicht nur unter toxikologischen, sondern auch unter sozioökonomischen Gesichtspunkten eine neue Dimension. Die jährlichen Kosten durch alkoholbedingte Lebererkrankungen in der BRD werden auf 4, 4 Mrd. DM geschätzt. Auch eine Vielzahl von Medikamenten kann ein breites Spektrum von Lebererkrankungen auslösen, die von Leberzellnekrosen bis zu Lebertumoren reichen. Vor der Verordnung von Schlafmitteln sollte an eine subklinische hepatische Encephalopathie als Auslöser gedacht werden. Der Zusammenhang zwischen geistig-seelischen Funktionen und der Leber wurden bereits von den Babyloniern und im alten China um die Jahrtausendwende vor Christi angenommen. Hippokrates beschrieb ein delirantes Zustandsbild, vermutlich eine fulminante Hepatitis. Galen mißt in seiner “Säftelehre” einer gestörten Lebersekretion eine große Bedeutung für psychopathologische Veränderungen zu. Erst im 18.Jahrhundert begannen eingehende Beschreibungen und Untersuchungen der neuropsychatrischen Symptomatik bei Lebererkrankungen.
Die Problematik zentralnervöser Funktionsveränderungen infolge alkohol- und schadstoffinduzierter Leberschädigungen gewinnt unter den Schätzungen der Deutschen Hauptstelle für Suchtgefahren, nach der ca. 5 % aller Bundesbürger suchtkrank sind und 6,6 Millionen Bundesbürger einen problematischen Alkoholkonsum pflegen, nicht nur unter toxikologischen, sondern auch unter sozioökonomischen Gesichtspunkten eine neue Dimension. Die jährlichen Kosten durch alkoholbedingte Lebererkrankungen in der BRD werden auf 4, 4 Mrd. DM geschätzt. Auch eine Vielzahl von Medikamenten kann ein breites Spektrum von Lebererkrankungen auslösen, die von Leberzellnekrosen bis zu Lebertumoren reichen. Vor der Verordnung von Schlafmitteln sollte an eine subklinische hepatische Encephalopathie als Auslöser gedacht werden. Der Zusammenhang zwischen geistig-seelischen Funktionen und der Leber wurden bereits von den Babyloniern und im alten China um die Jahrtausendwende vor Christi angenommen. Hippokrates beschrieb ein delirantes Zustandsbild, vermutlich eine fulminante Hepatitis. Galen mißt in seiner “Säftelehre” einer gestörten Lebersekretion eine große Bedeutung für psychopathologische Veränderungen zu. Erst im 18.Jahrhundert begannen eingehende Beschreibungen und Untersuchungen der neuropsychatrischen Symptomatik bei Lebererkrankungen.
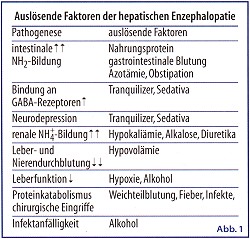
Die hepatische Encephalopathie kann im Gefolge einer Arzneimittel-induzierten chronischen Hepatopathie (meist Leberzirrhose) auftreten. (Abb. 1) Weitere Auslöser des Coma hepaticum sind Infektionen mit hepatogenen Viren, wie Hepatitis A, B, C und E, Delta Virus, Superinfektionen,Herpes-simplex und Coxsackie-Viren, Toxine wie Tetrachlorkohlenstoff, gelber Phosphor, Amanita phalloides, akute Alkohol-Hepatitis, sonstige Ursachen wie Morbus Wilson, Reye-Syndrom und Schwangerschaftsfettleber.
Das Erscheinungsbild der hepatischen Encephalopathie ist komplex und reicht von neuromuskulären Veränderungen zu zerebralen Funktionsstörungen.
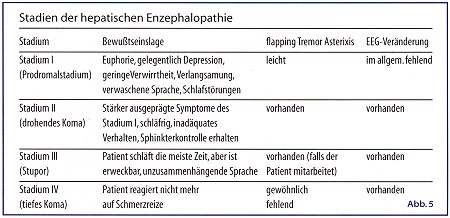
- Störungen der Feinmotorik mit Veränderungen des Schriftbildes und Asterixis, verwaschene Sprache. Feststellbar ist bei moderater HE Hyporeflexie, welche bei fortschreitendem HE in Hyperreflexie übergeht.
- Veränderung der Bewußtseinslage: Schlafstörungen, rasche Ermüdbarkeit, Verlangsamung und Lethargie, Desorientiertheit.
- Verschlechterung der intellektuellen Leistungsfähigkeit, Konzentrationsschwäche, Beeinträchtigung des Reaktionsvermögens, Nachlassen einfacher Rechenleistungen, Amnesie.
- Änderung des Verhaltens sehr wechselnder Ausprägung, Stimmungslabilität mit häufigem Überwiegen euphorischer Haltung in Verbindung mit Distanzlosigkeit, Geschwätzigkeit, Hemmungslosigkeit, Hang zur Selbstüberschätzung, im Verlauf dann ängstlich dysphorische Grundstimmung, paranoide Zustände im Präkoma.
Schlafstörungen als Symptome subklinischer Encephalopathien
Patienten mit Leberzirrhose weisen oft eine Veränderung des Schlaf-Wach-Rhythmus auf, die charakterisiert sind durch Einschlafstörungen in der Nacht und Somnolenz am Tag. In jüngster Zeit wurde dem Melatonin (N-Acetyl-5-Methoxytryptamin) neben zahlreichen anderen Wirkungen ein phasenregulierender Effekt im zirkadianen Schlaf-Wach-Rhythmus zugeordnet. Ein endogenes System vermittelt die Anpassung gemäß einer inneren Uhr an den Tag-Nacht-Rhythmus, wobei Photorezeptoren in der Retina Licht in elektrochemische Impulse umwandeln. Dabei scheint der suprachiasmatische Kern im vorderen Hypothalamus der zirkadiane”Pacemaker” zu sein, der die Melatoninausschüttung in der Nacht steuert. Schlafstörungen korrelieren mit Störungen des Melatoninstoffwechsels.Melatonin wird in der Hypophyse durch N-Acetylierung und 0-Methylierung aus Serotonin synthetisiert, wobei die N-Acetyltransferase der geschwindigkeitsbestimmende Schritt der Melatoninsynthese ist. Die nächtliche Aktivierung des Enzyms wird durch Freisetzung von Norepinephrin gesteuert. Die Eliminationshalbwertzeitvon Melatonin ist mit 45 Minuten kurz, wobei Melatonin durch Cytochrom-P450 hydroxyliert,an Sulfat gebunden und als 6-Sulfofhydroxymelatonin ausgeschieden wird. In einer Studie konnte durch radioaktive Markierung nachgewiesen werden, daß die Eliminationshaibwertzeit von Melatonin bei Patienten mit Leberzirrhose verlängert ist. Messungen des Melatoninspiegels in dem Patientenkollektiv alle 4 bis 6 Stunden ergaben erhöhte Melatoninspiegel sowohl zurTages-ais auch zur Nachtzeit infolge einer reduzierten hepatischen Clearance.Andere Autoren sehen einen direkten Einfluß von Ammoniak im Rahmen einer hepatischen Encephalopathie auf die Hypophyse. Ferner wird die Ammoniak-induzierte Erhöhung der Aufnahme von Tryptophan, der Serotonin-Vorstufe, im Gehirn als Auslöser eines modifizierten Melatoninstoffwechsels diskutiert. Bei Zirrhose-Patienten verlängert sich die Dauer des Melatonin-Outputs und das Melatonin-Maximum mit dem Korrelat eines veränderten Schlaf-Wach-Rhythmus.
Pathogenese der hepatischen Encephalopathie
Drei Hypothesen zur Pathogenese der hepatischen Encephalopathie werden derzeit diskutiert:
1. Wirkung endogener Neurotoxine
2. Wirkung falscher Neurotransmitter
3. Quantitative Veränderung der normalen Neurotransmitter und ihrer Rezeptoren
Endogene Neurotoxine
Als endogene neurotoxisch wirkende Substanzen bei hepatischer Encephalopathie kommen in Betracht:
– Ammonium
– Mercaptane
– kurz- und mittelkettige Fettsäuren
– Phenole
Die Ammoniumtherapie stellt die älteste Hypothese über die Pathogenese der hepatischen Encephalopathie dar.Vor mehr als 50 Jahren wurde beobachtet, daß sich nach Gabe von Ammoniak bei Patienten mit Leberzirrhose ein Koma entwickelte.
Die Hyperammoniumämie bei Lebererkrankungen beruht auf einer verminderten Ammoniumentgiftung in der Leber. die Verminderung kann hämodynamisch durch Ausschaltung der Leber aus dem Pfortaderkreislauf infolge Zirrhose und Ausbildung eines Kollateralkreislaufs verursacht sein. Toxische Substanzen, die im Darm gebildet werden, vor allem Ammoniak und Eiweißabbauprodukte gelangen unter Umgehung der Leber in den großen Kreislauf und somit unentgiftet ins Gehirn. Eine weitere Ursache der Hyperammoniumämie ist metabolisch begründet aufgrund einer Einschränkung der Harnstoff- und Glutaminsynthese.
Durch den Aminosäurestoffwechsel entsteht in vielen Organen Ammoniak, das schon in relativ niedrigen Konzentrationen als Zellgift wirkt. Die notwendige Umwandlung von Ammoniak in weniger toxische Stoffwechselprodukte erfolgt in erster Linie über die Bildung von Glutamin aus Ammoniak und Glutaminsäure, katalysiert durch die Glutamin-Synthetase. Dem Organismus steht ein weiterer Weg zur Entgiftung des Ammoniaks über die Bildung von Harnstoff in der Leber zur Verfügung.
Das Gehirn besitzt auch zwei Wege, um Ammoniak zu entgiften. Zum einen ist es derzitierte, in Form von Glutamin aus Glutamat. Dieser Entgiftungsweg ist bei normaler Hirnfunktion ausreichend. Steigende Ammoniakwerte können nicht über diesen Mechanismus eliminiert werden. Der zweite Weg ist die reduktive Eleminierung über c-Ketoglutarat zu Glutamat. Dieser Abbauweg wird nur bei steigenden Ammoniakwerten eingeschlagen. Glutamin ist der Precursor sowohl für Glutamat als auch für GABA (GammaAminobuttersäure),welche in praesynaptischen Neuro nen synthetisiert werden.
Nach Freisetzung der Transmitter in den synaptischen Spalt wird die Aktivität durch Aufnahme in die Glia-Zellen modifiziert. In den Glia-Zellen wird GABA zu Glutamat umgewandelt. Dieser Glutamat-Glutamin-Glutamat-Zyklus zwischen Glia-Zellen und Neuronen kann durch Ammonium beeinflußt werden. Ammoniak kann die neuronale Glutaminase blockieren. Man nimmt an, daß durch mehrere Mechanismen eine Hyperammoniumämie die erregende Neurotransmission im ZNS supprimieren kann. Eine zweite Hypothese über die Auswirkungen einer Hyperammoniumämie beschreibt eine Beeinflussung des cerebralen Metabolismus. Dabei soll es über die Wechselwirkung des Malat-Aspartat-Austausches zu einer Abnahme des cerebralen ATP-Gehaltes kommen. Diese Hypothese wird durch die Beobachtung unterstützt,daß bei Tieren mit portokavalen Shunts unter Belastung der Laktat/Pyruvatquotient zunimmt, die Konzentration von energiereichem Phosphat (ATP) abnimmt. Die dritte Hypothese beinhaltet den Einfluß von Ammoniak auf die Blut-Liquor-Schranke. So stimuliert Ammoniak die Aufnahme aromatischer Aminosäuren (insbesondere Tryptophan). Ferner hat eine Hyperammoniumämie einen Einfluß auf postsynaptische Rezeptoren. Die Dichte der Serotoninrezeptoren nimmt ab, ihre Affinität zu. Ein weiterer Ansatzpunkt zur Pathogenese der Amoniak-induzierten Neurotoxizität sind Effekte auf die neuronale Elektrophysiologie. Ammoniak erhöht das Ruhemembranpotential und hemmt die axonale Leitfähigkeit und das exzitatorische postsynaptische Potential.
 Weitere Neurotoxine
Weitere Neurotoxine
Mercaptane
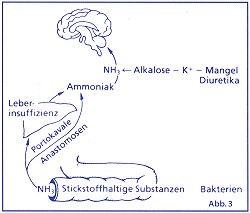
Die Konzentration von neurotoxischen Mercaptanen Methantiol, Dimethylsulfid und Äthylmercaptan in Blut, Liquor und Urin sind bei Patienten mit Leberzirrhose und hepatischer Encephalopathie erhöht. Sie entstehen beim bakteriellen Abbau der Aminosäure Methionin. Ihre Neurotoxizität wird auf die Hemmung der Na-KATPase zurückgeführt. Mercaptane in der Atemluft verursachen wahrscheinlich den Foetor hepaticus. Durch Methioninzufuhr kann eine hepatische Encephalopathie ausgelöst werden. Bei einer Leberzirrhose steigen die GABA-Spiegel im Blut durch funktionelle und anatomische Shunts an. (Abb. 3)
Neben der erhöhten GABA-Konzentration im Gehirn führt eine Zunahme der GABA-Rezeptoren zu einer Neurodepression. Serotonin vermittelt als Neurotransmitter überspezifische Rezeptoren eine gesteigerte Schlafbereitschaft und erniedrigte motorische Aktivität.Außerdem spielt Serotonin bei dem Appetit- und Aggressionsverhalten eine Rolle. Ammonium wird im Gehirn in den Astrozyten durch vermehrte Bildung von Glutamin entgiftet. Glutamin wird an der Blut-Liquor-Schranke gegen neutrale Aminosäuren, insbesondere aromatische, ausgetauscht.
Die Pathogenese der hepatischen Encephalopathie ist also ein multifaktoreller Prozeß, an dem mehrere Mechanismen und Toxine synergistisch beteiligt sind. (Abb.4)
 Hepatische Biotin- und Vitamin-Bl2-Speicherstörungen als Auslöser einer Propionyl- bzw. Methylmalonylacidurie mit dem Korrelat einer Hyperammoniämie
Hepatische Biotin- und Vitamin-Bl2-Speicherstörungen als Auslöser einer Propionyl- bzw. Methylmalonylacidurie mit dem Korrelat einer Hyperammoniämie
Biotin
Schon seit längerem ist bekannt, daß bei schlechtgenährten Alkoholikern mit Leberschädigung in etwa 40 % der Fälle niedrige Biotin-Spiegel zu finden sind. Bei bestimmten Hepatopathien sind die Serum-Biotinspiegel reduziert, bei Leberzirrhose um etwa 81 %,beim Fettlebersyndrom um etwa 26%. Leberzirrhose scheint die Utilisation und Speicherung von Biotin zu verhindern.Auch Hautveränderungen wie Erythrosis interfollic. collic Sklerenikterus, teleangiektatische Gesichtsröte, Lackzunge und Lacklippen werden als Korrelate veränderter Zink- und Biotinspiegel im Plasma bei chronischem Alkoholismus diskutiert. Alopezie, Ataxie und Keratokonjunktivitis können bei Biotin-bedingten Enzymdefekten auftreten. Interessant ist in diesem Zusammenhang, daß ein angeborener Mangel der Biotin-abhängigen Propionyl-CoA-Carboxylase,von dem in der internationalen medizinischen Literatur mehrere Dutzend Fälle beschrieben worden sind, die Symptomatikeinerschweren Ketoacidose, Erbrechen, Hyperventilation und schwerer neurologischer Ausfälle aufweist. Laborchemisch läßt sich eine metabolische Azidose, eine Hyperammoniämie sowie eine erhöhte Propionsäure-Konzentration nachweisen.
Hyperammoniämie als Korrelat der Störung des Biotin-abhängigen Propionsäurestoffwechsels
Die Biotin-abhängige Propionyl-CoA-Carboxylase vermittelt den Abbau ungeradzahliger Fettsäuren und mancher Aminosäuren,wie z. B.Isoleucin,Valin. Methionin und Threonin. Patienten mit einem Propionylcarboxylase-Mangel akkumulieren Propionat und exkretieren ferner große Mengen an Methylcitrat, 3-Hydroxypropionat und Propionylglycin in Harn. lnteressanterweise gibt es beträchtliche Variationen der klinischen Expression der Propionazidämie, indem manche Patienten mit angeborenem Propionyl-CoA-Carboxylase-Mangel nur wenige Symptome, andere jedoch alle klassischen Symptome der Erkrankung aufweisen.
Für die Pathogenese der hepatischen Enzephalopathie besitzt das Phänomen der Propionyl-CoA-Azidämie Relevanz, da sie in den Harnstoffzyklus eingreifen und die Ammoniakeliminierung modifizieren kann. Propionyl CoA hemmt in Rattenmitochondrien die N-Acetylglutamat-Synthetase signifikant, die ihrerseits die Carbamoylphosphatsynthetase induziert. Sowohl in Homogenisaten aus Ratten- und Menschenleber konnte für Propionyl CoA und in einem geringeren Ausmaß fürTiglyl-CoA und Methylmalonyl-CoA ein zeit- und konzentrationsabhängiger Hemmeffekt auf die Carbamoylphosphatsynthetase nachgewiesen werden.
Interessant erscheint in diesem Zusammenhang die Theorie, daß Antikonvulsiva wie Phenytoin, Primidon, Phenobarbital und Carbamazepin über eine Interaktion mit dem Biotinstoffwechsel hemmende Neurone aktivieren bzw. durch Akkumulation von C02 antikonvulsiv wirken. Tatsächlich wurde bei über 80 % von 117 antikonvulsiv behandelten Patienten eine Erniedrigung des Biotinspiegels im Plasma gefunden.Signifikante negative Korrelationen zeigten sich zur durchschnittlichen Tagesdosis und zur Gesamtmenge eingenommener Antiepileptika sowie zum Phenytoin-Spiegel im Plasma. Diskutiert werden eine Akkumulation von C02 sowie eine Senkung von Aspartat im Hirngewebe als Mediatoren des antikonvulsiven Effektes sowie eine Erhöhung des inhibitorischen Neurotransmitter Glycin über einen Propionyl-Carboxylase-Mangel. Es drängt sich die Assoziation auf, daß Arzneimittel-induzierter Biotinmangel mit dem Korrelat einer Hyperammoniämie Vehikel zur Erhöhung des Ruhemembranpotentials und damit derantikonvulsiven Wirkung sein könnte.Auch Nebenwirkungen von Antiepileptika wie zerebelläre störungen und Dermatitiden sind Korrelate eines veränderten Biotinstoffwechsels. Chauhan und Dakshinamurti konnten 1988 nachweisen, daß Pheno- und Pentobarbital sowie Carbamazepin und Phenytoin kompetitiv um das Bindungsprotein Biotinidase konkurrieren.
Vitamin B12
In Ratten mit Vitamin-B12-Mangel verursachte eine Methylmalonacidurie einen um den Faktor 15 erhöhten Propionyl-CoA-Leberspiegel. Methylmalonsäure ist Produkt der Biotin-abhängigen Propioylcarboxylase, die im intermediären Stoffwechsel über die Vitamin-B12-abhängige MethylmalonylCoA-Mutase in Succinyl-CoA abgebaut wird. Daraus kann gefolgert werden, daß ein Vitamin-B12-Mangel direkt, da auch Methylmalonyl-CoA in Konzentrationen von 3 mMol eine 30- bis 70prozentige Hemmung der N-Acetylglutamatsynthase verursacht, als auch indirekt über eine Propionacidämie eine reduzierte Harnstoffbildung über den Harnstoffzyklus mit dem Korrelat einer Hyperammoniämie verursacht. Lebererkrankungen können die Speicherfähigkeit für Vitamin B12 in erheblicher Weise hemmen. In der Leber erfolgt die Speicherung und Umwandlung der aktiven Coenzyme Methylcobylamin und 5-Desoxyadenosylcobalamin, Äthylalkohol und sein Abbauprodukt Acetaldehyd interagieren mit Glutathion und senken den Pool übertragbaren Methylgruppen in Form des Adenosylmethionins (siehe Neurodate 3/98). Adenosylmethionin ist erforderlich zur Methylierung des Cobalamins.
Therapie der hepatischen Encephalopathie
Bei der hepatischen Encephalopathie gilt der Suche nach einem potentiellen Auslöser primäres Interesse, um diese eliminieren zu können.
Dazu gehören – nach Häufigkeit:
– gastrointestinale Blutungen
– massive Diuretikatherapie (mit den Komplikationen der Hyperkaliämie und Alkalose)
– zu hohe alimentäre Proteinzufuhr
– Infekte
– Alkoholexzeß
– Sedativa/Hypnotika
Am häufigsten tritt die hepatische Encephalopathie nach gesteigerter Proteinaufnahme, nach gastrointestinalen oder Weichteilblutungen oder Azotämie auf. Unter diesen Bedingungen kommt es zu einer vermehrten Bildung stickstoffhaltiger Metaboliten. Das bei ihrem Abbau entstehende Ammoniak wird von der insuffizienten Leber unzureichend entgiftet. (Abb. 5) Gastrointestinale Blutungen sind häufig von Hypovolämien, Hypoxie und Schock begleitet, die ihrerseits die Leberdurchblutung und die Ammoniakelimination verschlechtern. Störungen des Flüssigkeits- und Elektrolythaushalts mit Hypovolämien, Hypernatriämien, Hypokaliämien und Alkalose sowie Diuretikatherapie können in etwa 50 % der Fälle eine hepatische Encephalopathie auslösen. Die Ursache ist eine gesteigerte renale Ammoniumproduktion, eine verminderte Leber- und Nierendurchblutung sowie eine Hemmung der Harnstoffsynthese durch einige Diuretika. Bei klinisch manifester Symptomatik,also ab Grad 1 einer Encephalopathie, ist die diätetische Beratung Therapiegrundlage. Entsprechend der Proteintoleranz werden Ernährungspläne mit eingeschränkter Eiweißzufuhr aufgestellt. Dabei ist der Ernährungszustand zu berücksichtigen, der bei chronisch Leberkranken in der Mehrzahl unzureichend ist. Dies ist bei der Proteinzufuhr zu bedenken, denn mit etwa 50 g Eiweiß/Tag kann zwar noch eine ausgeglichene Stickstoffbilanz erzielt werden; aber wenn Mangelzustände vorliegen, reicht diese Menge nicht aus,um Defizite auszugleichen, so daß dann eine höhere Eiweißzufuhr anzustreben ist. Dabei läßt sich die Proteintoleranz durch verschiedene Maßnahmen verbessern. Bevorzugung pflanzlichen Proteins und faserreiche Kost sowie gleichmäßige Verteilung der Nahrungszufuhr über den Tag können zunächst versucht werden. Kontinuierliche Lactulosetherapie (Bifiteral,Lactofalk, Lactuflor, Lactulose Neda, Lactulose Hek), die entsprechend der Defäkationsfrequenz, die 2- bis 3mal/Tag liegen sollte, dosiert wird, stellt die nächste Maßnahme dar.
Lactulose
Lactulose ist ein synthetisches Disaccharid (1, 4-b-Galaktosidfruktose), welches im Dünndarm weder resorbiert noch gespalten,aber im Dickdarm durch die Bakterienflora zu Milchsäure abgebaut wird. Lactulose führt deshalb zu einer Ansäuerung des Darminhalts, die ihrerseits durch Laxation, Veränderung des bakteriellen Stoffwechsels und durch Ammoniumverteilung nach dem Ph-Gradienten die Ammoniumresorption aus dem Kolon hemmt. Der günstige Effekt von Laktulose in der Therapie der hepatischen Encephalitis ist gleich zusetzen mit dem der nicht resorbierbaren Antibiotika. Mit ähnlichem Erfolg kann man anstelle von Lactulose auch Laktilol (b-Galaktosido-Sorbitol) verwendet werden. Kurz- und mittelkettige Fettsäuren entstehen aus Nahrungsfetten unter Einwirkung von Darmbakterien und bei schweren Lebererkrankungen durch den unkompletten Abbau der normalen Fettsäuren.
 Phenole werden ebenfalls durch Mikroorganismen im Darm gebildet. Die Neurotoxine verhalten sich synergistisch. Es besteht keine enge Korrelation zwischen der Konzentration eines einzelnen Neurotoxins und der Manifestierung einer hepatischen Encephalopathie.
Phenole werden ebenfalls durch Mikroorganismen im Darm gebildet. Die Neurotoxine verhalten sich synergistisch. Es besteht keine enge Korrelation zwischen der Konzentration eines einzelnen Neurotoxins und der Manifestierung einer hepatischen Encephalopathie.
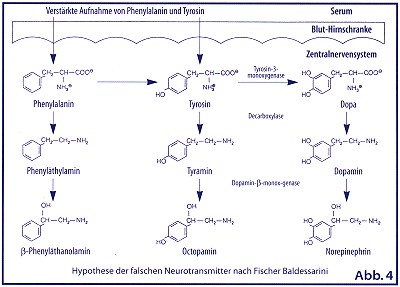
Bei einer Leberzirrhose führt die nachweisbare Aminosäureninbalance im Serum (Abnahme der verzweigtkettigen Aminosäuren/Zunahme der aromatischen Aminosäuren) zu einem gesteigerten Transport von Phenylalanin, Tyrosin und Tryptophan durch die Blut-Liquor-Schranke. Dieser Vorgang wird durch die Hyperammoniumämie weiter verstärkt. Da Phenylalanin in höheren Konzentrationen dieTyrosin-3-Monooxygenase hemmt, kommt es zur Bildung falscher Transmitterstoffe, wie Octopamin, Phenaläthyamin und Phenyläthanoamin, die im Vergleich zu den normalen Neurotransmittern Dopamin, Norepinephrin nur einen Bruchteil (ca. 1/50) der neuroexzitatorischen Aktivität besitzen. (Abb. 1)
Gleichzeitig wird aus Tryptophan vermehrt der inhibitorische Neurotransmitter Serotonin gebildet . Auch die Veränderungen normaler Neurotransmitter und ihrer Rezeptoren sind von Bedeutung. Gamma-Amino-Buttersäure ist neben Glycin der wichtigste inhibitorische Neurotransmitter im Gehirn. Der GABA-Rezeptor ist identisch mit dem Diazepam- und Babituratrezeptor. GABA wird auch durch Darmbakterien gebildet, aber in der Leber normalerweise vollständig abgebaut.(Abb.2)
Antibiotika
Intestinal weitgehend nicht resorbierbare Antibiotika (Paromomycin, Neomycin) wurden erfolgreich zur Therapie der hepatischen Encephalopathie eingesetzt. Ziel ist es dabei, durch Reduktion der Bakterien die Ammoniumproduktion im Darm zu senken. Obwohl nur Bruchteile der eingesetzten Antibiotika resorbiert werden, besteht dennoch die Gefahr der Oto- und Nephrotoxizität. Sie sollten daher nur kurzfristig angewendet werden.
Der Hauptabbauweg des Ammoniaks läuft über den ausschließlich in der Leber stattfindenden Harnstoffzyklus. Ziel war es daher, durch die Gabe von Substraten des Harnstoffzyklus diesen und den Zitronensäurezyklus zu stimulieren und die Ammoniakentgiftung zu fördern. Eingesetzt wurden dafür Argininmalat und Ornithinaspartat. Interessant dürften insbesondere die Ergebnisse einer Kombinationstherapie mit Lactulose sein.
Das geschwindigkeitsbestimmende Enzym des Harnstoffzyklus, das zugleich Ammonium in den Zyklus einschleust, ist die Carbamoylphosphatsynthetase. Verzweigtkettige Aminosäuren hemmen das Enzym für den Abbau des Ornithins und stimulieren wahrscheinlich die N-Azetyl-Synthetase, so daß auf beiden Wegen die Ammoniumentgiftung im Harnstoffzyklus gesteigert wird.
Verzweigtkettige Aminosäuren
Verzweigtkettige Aminosäuren mit einem hohen Anteil an Leucin, Isoleucin und Valin haben eine Wirksamkeit bei der hepatischen Encephalopathie. Der Effekt liegt in einer Verbesserung der hepatischen Ammoniumentgiftung. Verzweigtkettige Aminosäuren werden vorwiegend in der Muskulatur und im Fettgewebe abgebaut, hemmen den Proteinabbau in Leber und Muskulatur und stimulieren die Proteinsynthese.
Begriffsdefinitionen
Encephalopathie:
Sammelbegriff für nichtentzündliche Erkrankungen oder Schädigungen des Gehirns
Leberzirrhose:
diffuse chronische Lebererkrankung; infolge Parenchymuntergang kommt es zu bindegewebiger Narbenbildung. Umgestaltung des Gefäßapparates und regeneratorischem Parenchymumbau.
Shunt, portokavaler:
Operative Herstellung einer Verbindung zwischen dem Pfortadergebiet und derVena cava bei portaler Hypertension (blutende Oesophagusvarizen)
Foetor hepaticus:
charakteristischer Mundgeruch nach frischer Leber oder Lehmerde bei Leberzerfall (Leberdystrophie, akute gelbe Leberatrophie, Leberkoma durch Leberzerfall
Hypovolämie:
Verminderung der zirkulierenden Blutmenge durch Verlust von Vollblut, Plasma Albumin und Wasser. Ursachen: Blutverlust nach außen, in Körperhöhlen oder Gewebe nachTraumen,akuten Erkrankungen,z,B, Ulkus, Ösophagusvarizenblutungen. Plasmaverluste nach Verbrennungen, Flüssigkeitsverluste bei akuten Erkrankungen.
Azotämie:
Vermehrung des Harnstoffs und harnpflichtiger N-Substanzen im Blut bzw. Harn
Hypoxie:
Sauerstoffmangel, Sauerstoffnot, Herabsetzung des P02 im Blut bzw. Körpergeweben. Symptomenkomplex aus Lufthunger mit Atemnot, Unruhe, Angstzuständen und Verwirrtheit,Tachykardie und Blutdruckanstieg.
Asterixis:
syn. Flapping Tremor: Wackeltremor, Unfähigkeit der Hände infolge intermittierenden Tonusverlustes eine bestimmte Haltung einzunehmen
Gliazellen:
Neuroglia-Stützgewebe des Zentralnervensystems das ein dreidimensionales Faserwerk bildet, in das die Nervenzellen und ihre Fortsätze eingeschlossen sind, grenzt die nervöse Substanz an allen Oberflächen und gegen die Blutgefäße ab. Ist für den Stoffwechsel des Nervengewebes von großer Bedeutung.
Jens Bielenberg, (Apotheker)
zurück zur Übersicht dieser Ausgabe
 Wir beraten Sie gerne
Wir beraten Sie gerneHier geht's zur Paracelsus Schule Ihrer Wahl.
